Science always keeps me busy and entertained!
Watch this space! I am currently studying intricate relationship between myelin and sleep...
Myelination sychronize cortical oscullations by consolidating parvalbumin-mediate phasic inhibition. (Elife, 2022)

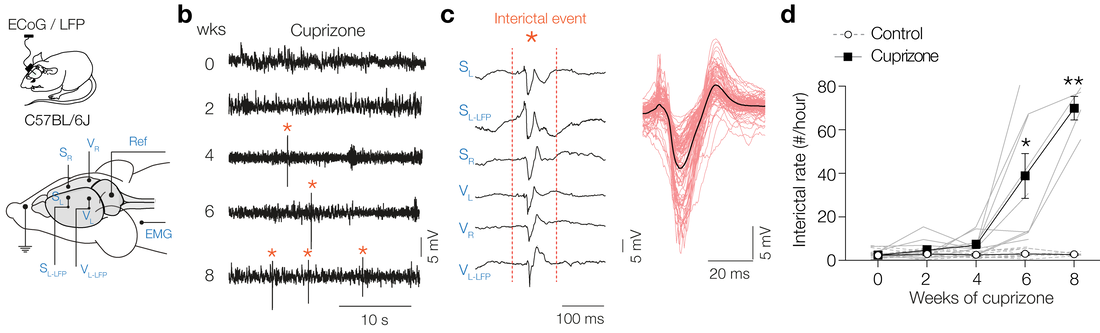
Progressive demyelination causing interictals
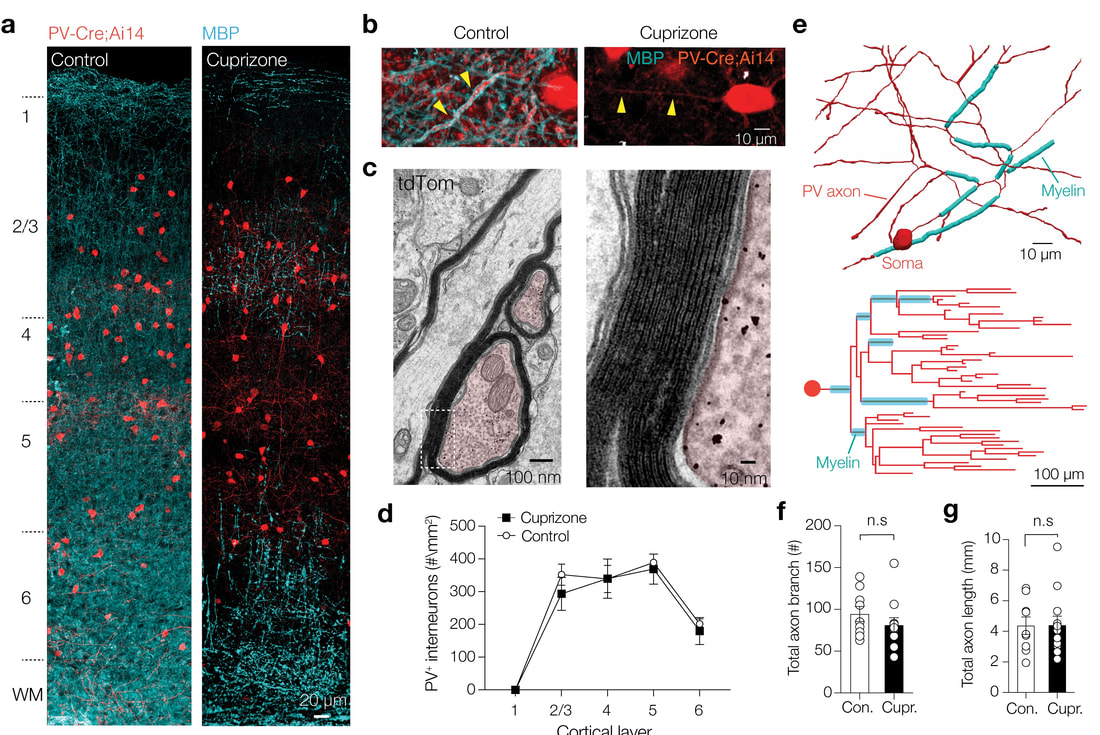
progressive demyelination do affect the PV-axon morphology
Link to the study:
elifesciences.org/articles/73827 |

Parvalbumin-positive (PV+) γ-aminobutyric acid (GABA) interneurons are critically involved in producing rapid network oscillations and cortical microcircuit computations but the significance of PV+ axon myelination to the temporal features of inhibition remains elusive. Here using toxic and genetic mouse models of demyelination and dysmyelination, respectively, we find that loss of compact myelin reduces PV+ interneuron presynaptic terminals, increases failures and the weak phasic inhibition of pyramidal neurons abolishes optogenetically driven gamma oscillations in vivo. Strikingly, during behaviors of quiet wakefulness selectively theta rhythms are amplified and accompanied by highly synchronized interictal epileptic discharges. In support of a causal role of impaired PV-mediated inhibition, optogenetic activation of myelin-deficient PV+ interneurons attenuated the power of slow theta rhythms and limited interictal spike occurrence. Thus, myelination of PV axons is required to consolidate fast inhibition of pyramidal neurons and enable behavioral state-dependent modulation of local circuit synchronization.
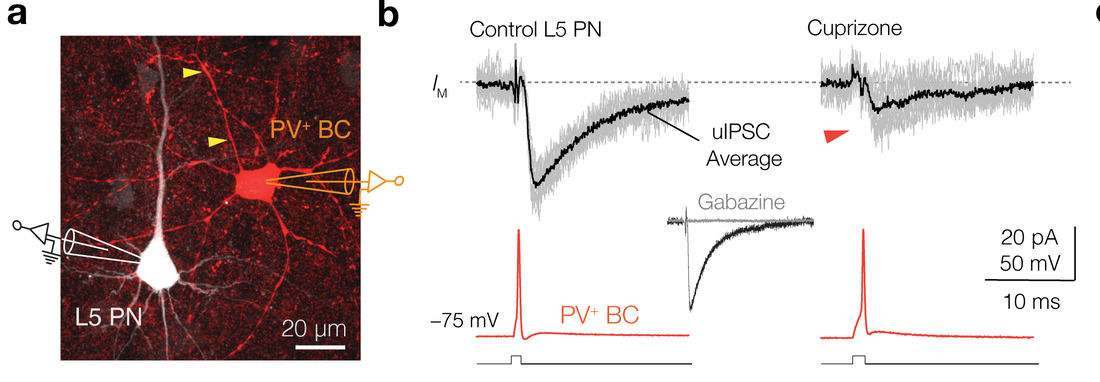
Loss of PV-phasic inhibition without affecting condution velocity
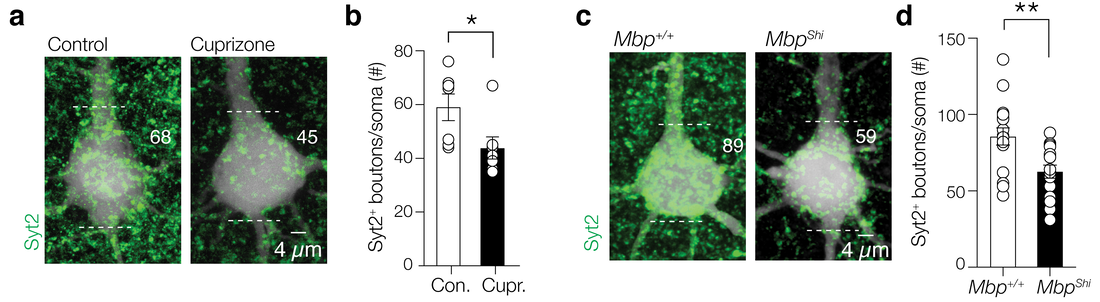
Loss of per-synaptic inhibitory boutons
|
Complement-associated loss of CA2 inhibitory synapses in the demyelinated hippocampus impairs memory. (Acta Neuropathologica, 2021)

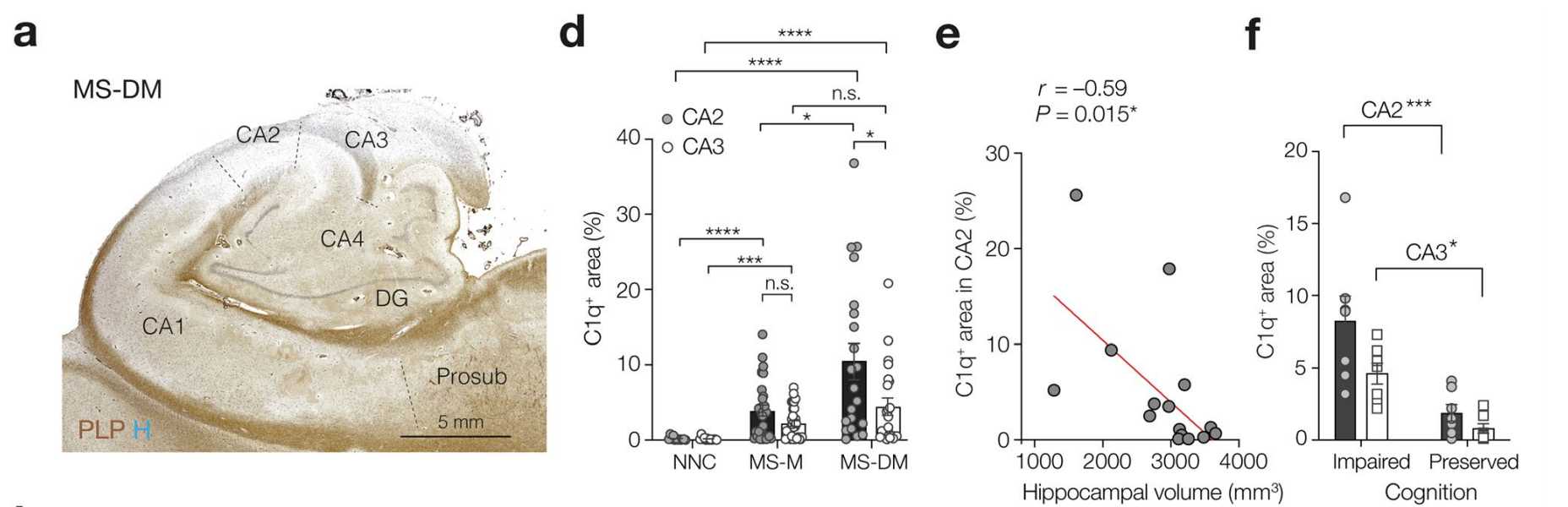
Myelin loss in human hippocampus correlated with cognition impairments
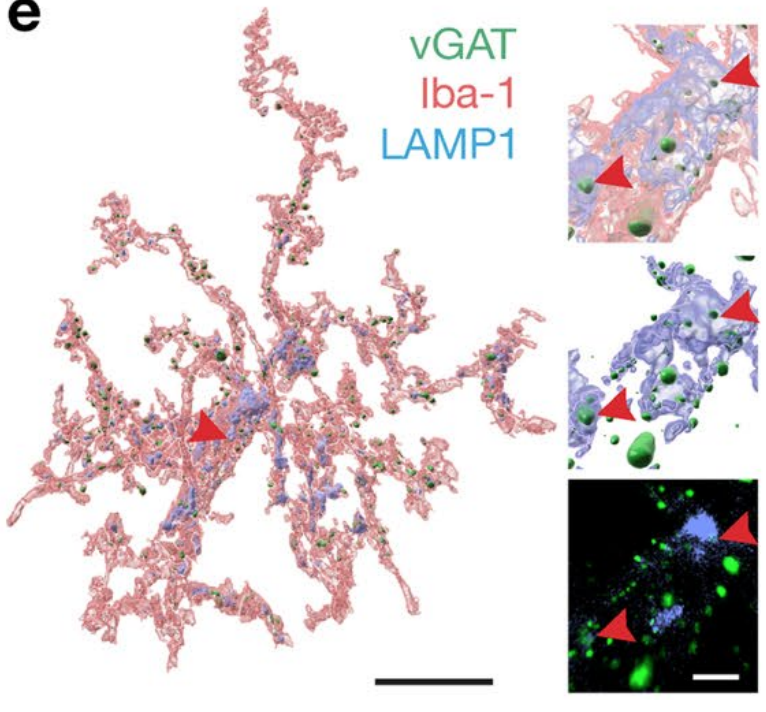
Microglia (immune cells in the brain, iba-1) eating inhibitory synapses (green) in the hippocampus.
|
The complement system is implicated in synapse loss in the MS hippocampus, but the functional consequences of synapse loss remain poorly understood. Here, in post-mortem MS hippocampi with demyelination we find that deposits of the complement component C1q are enriched in the CA2 subfield, are linked to loss of inhibitory synapses and are significantly higher in MS patients with cognitive impairments compared to those with preserved cognitive functions. Using the cuprizone mouse model of demyelination, we corroborated that C1q deposits are highest within the demyelinated dorsal hippocampal CA2 pyramidal layer and co-localized with inhibitory synapses engulfed by microglia/macrophages. In agreement with the loss of inhibitory perisomatic synapses, we found that Schaffer collateral feedforward inhibition but not excitation was impaired in CA2 pyramidal neurons and accompanied by intrinsic changes and a reduced spike output. Finally, consistent with excitability deficits, we show that cuprizone-treated mice exhibit impaired encoding of social memories. Together, our findings identify CA2 as a critical circuit in demyelinated intrahippocampal lesions and memory dysfunctions in MS.
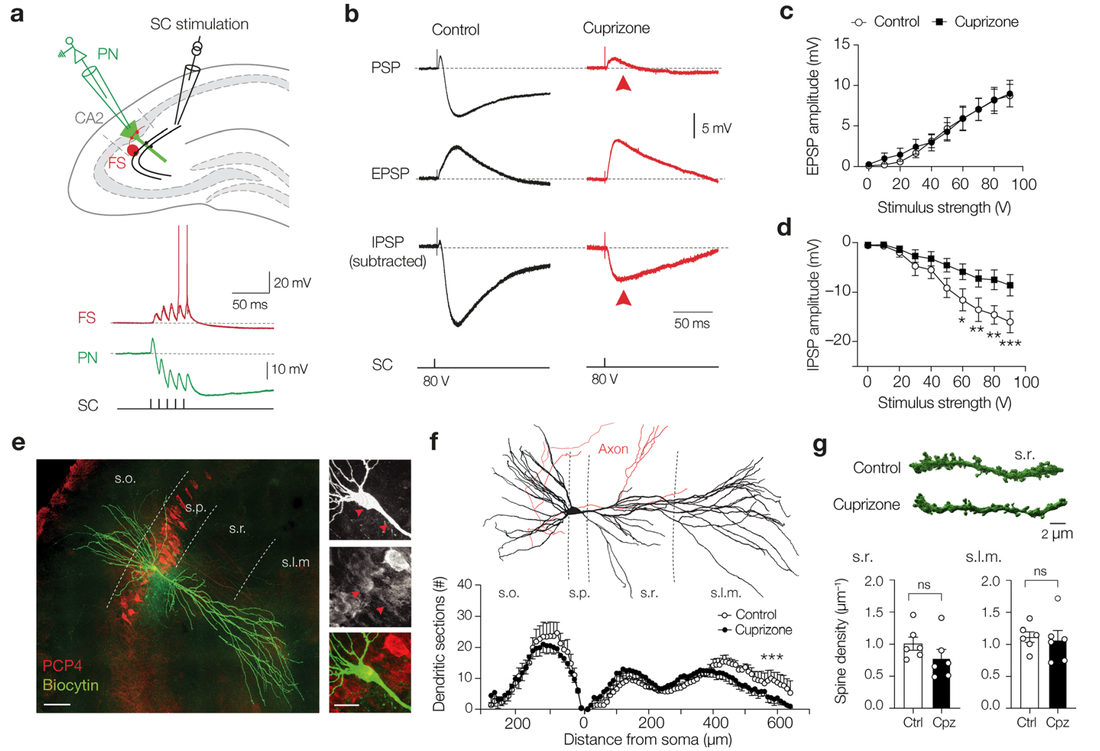
Loss of inhibitory signal in hippocampus CA2 microcircuit.
|
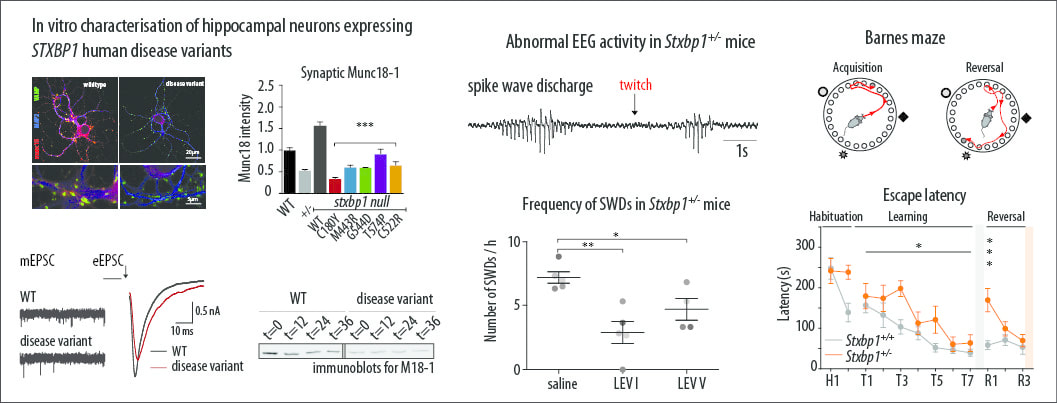
Protein instability, haploinsufficiency, and cortical hyper-excitability underlie STXBP1 encephalopathy. (Brain; 12-03-2018)

|
A collaboration and a comprehensive study about intellectual disability and recurrent seizures (epilepsy) in genetic childhood encephalopathy is online. My contribution was to teach the lead author to use radiowave transmission to track and monitor epilepsy! The study will appear in print in may at the journal Brain. De novo heterozygous mutations in STXBP1/Munc18-1 cause early infantile epileptic encephalopathies (EIEE4, OMIM #612164) characterized by infantile epilepsy, developmental delay, intellectual disability, and can include autistic features. We characterized the cellular deficits for an allelic series of seven STXBP1 mutations and developed four mouse models that recapitulate the abnormal EEG activity and cognitive aspects of human STXBP1-encephalopathy. Disease-causing STXBP1 variants supported synaptic transmission to a variable extent on a null background, but had no effect when overexpressed on a heterozygous background. All disease variants had severely decreased protein levels. Together, these cellular studies suggest that impaired protein stability and STXBP1 haploinsufficiency explain STXBP1-encephalopathy and that, therefore, Stxbp1+/− mice provide a valid mouse model. Simultaneous video and EEG recordings revealed that Stxbp1+/− mice with different genomic backgrounds recapitulate the seizure/spasm phenotype observed in humans, characterized by myoclonic jerks and spike-wave discharges that were suppressed by the antiepileptic drug levetiracetam. Mice heterozygous for Stxbp1 in GABAergic neurons only, showed impaired viability, 50% died within 2–3 weeks, and the rest showed stronger epileptic activity. c-Fos staining implicated neocortical areas, but not other brain regions, as the seizure foci. Stxbp1+/− mice showed impaired cognitive performance, hyperactivity and anxiety-like behaviour, without altered social behaviour. Taken together, these data demonstrate the construct, face and predictive validity of Stxbp1+/− mice and point to protein instability, haploinsufficiency and imbalanced excitation in neocortex, as the underlying mechanism of STXBP1-encephalopathy. The mouse models reported here are valid models for development of therapeutic interventions targeting STXBP1-encephalopathy. Complete study can be found at ; academic.oup.com/brain/advance-article/doi/10.1093/brain/awy046/4929905 |
Seizures and disturbed brain potassium dynamics in Leukodystrophy MLC
(Annals of Neurology; 21-02-2018)
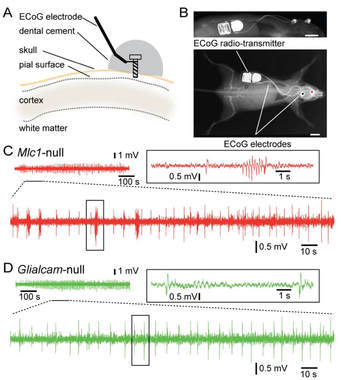
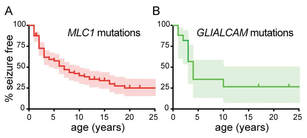
Epilepsy in MLC patients
Epilepsy monitoring in MLC mice using radio transmission
|
Loss of function of the astrocyte-specific protein MLC1 leads to the childhood onset leukodystrophy “megalencephalic leukoencephalopathy with subcortical cysts” (MLC). Studies on isolated cells show a role for MLC1 in astrocyte volume regulation and suggest that disturbed brain ion and water homeostasis is central to the disease. Excitability of neuronal networks is particularly sensitive to ion and water homeostasis. In line with this, reports of seizures and epilepsy in MLC patients exist. However, systematic assessment and mechanistic understanding of seizures in MLC are lacking.
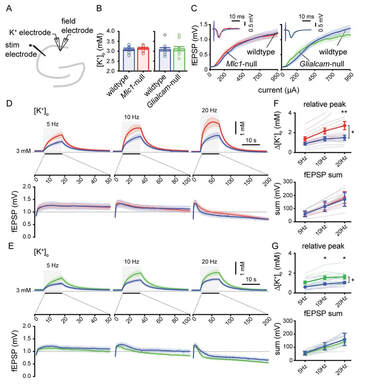
We analyzed an MLC patient inventory to study occurrence of seizures in MLC. We used two distinct genetic mouse models of MLC to further study epileptiform activity and seizure threshold through wireless extracellular field potential recordings. Whole cell patch-clamp recordings and K+-sensitive electrode recordings in mouse brain slices were used to explore the underlying mechanisms of epilepsy in MLC. An early onset of seizures is common in MLC. Similarly, in MLC mice we uncovered spontaneous epileptiform brain activity and a lowered threshold for induced seizures. At the cellular level, we found that although passive and active properties of individual pyramidal neurons are unchanged, extracellular K+ dynamics and neuronal network activity are abnormal in MLC mice. Disturbed astrocyte regulation of ion and water homeostasis in MLC causes hyperexcitability of neuronal networks and seizures. These findings suggest a role for defective astrocyte volume regulation in epilepsy. This article is protected by copyright. All rights reserved. Complete research study can be found at: Annals of Neurology; onlinelibrary.wiley.com/doi/10.1002/ana.25190/full |
Disturbed potassium dynamics in MLC mice hippocampus
Brain ion water homeostasis. Department of Child Neurology, VUMC and Department of Integrative neurophysiology, VU University, The Netherlands.
(PhD date; 14-11-2017)
|
The adult human brain consists for approximately 70% of water. The correct content and compartmentalization of brain water (or water homeostasis) are fundamental for its normal functioning. Unlike other body tissues, the brain is encased by a rigid bony skull, and even small increases in tissue volume produce significant rises in intracranial pressure, causing compression of neural tissue and damage, mostly due to reduced blood flow. If progressive and not successfully treated, brain herniation and death may follow. Only in infants, where the skull sutures have not closed yet, compensation through abnormal increase in head size may occur. Brain edema, increased tissue water content, may involve different compartments: extracellular, intracellular and intramyelinic. Brain edema is a dangerous and frequently life threatening neurological complication, and is an important but enigmatic research topic for basic scientific research and clinical medicine alike. Clinical neurologists most frequently observe symptomatic brain edema following head injuries and in connection with cerebral tumors. By contrast, rare types of chronic intramyelinic brain edema caused by monogenic defects are associated with more subtle clinical phenotypes with slower progression. Megalencephalic leukoencephalopathy with subcortical cysts (MLC) is a unique example of genetically determined white matter disease characterized by chronic intramyelinic brain edema.
|
The principal aim of the work presented in this thesis was to examine a genetic disease characterized by chronic white matter edema, megalencephalic leukoencephalopathy with subcortical cysts (MLC), in order to increase our understanding of this disease as well as that of water homeostasis in the healthy brain. Over the course of three chapters, I tested the validity of two mouse models for MLC (Mlc1-null and Glialcam-null mouse). Importantly, I then used these MLC mice to investigate the pathophysiological mechanisms leading to activity-dependent brain edema.
Second mouse model for human MLC disease. Department of Child Neurology, Amsterdam Neuroscience.
(Annals of Clinical and translational Neurology)
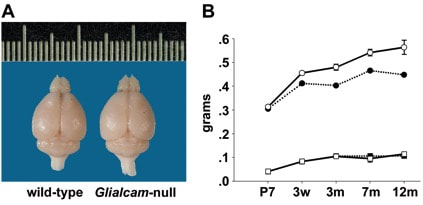
Megalencephaly with increased brain water content in Glialcam-null mice. (A) Glialcam-null mice have larger brains than wild-type littermates, as shown for these 8-month-old animals. (B) Measurements of brain wet and dry weight at postnatal day 7 (P7), 3 weeks (w), and at 3, 7, and 12 months, show significantly increased brain wet weight (circles) in Glialcam-null mice (solid line) compared to controls (dotted line) from 3 weeks
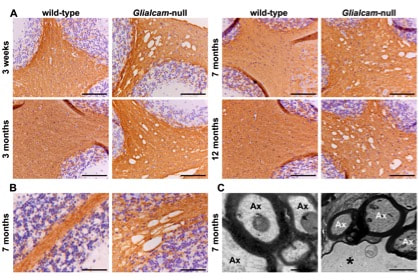
Normal myelination and intramyelinic edema in the white matter of Glialcam-null mice. (A) Stain for the myelin-specific protein proteolipid protein (PLP) of the cerebellum shows no differences in PLP staining intensity between Glialcam-null and wild-type white matter at all ages analysed, indicating that the mutant mice have no lack or loss of myelin. (B) Stain for PLP shows that the fine tissue strands crossing the vacuoles are PLP-positive. (C) Ultrastructural analysis of the cerebellar white matter of 8-month-old Glialcam-null mice shows large vacuoles surrounded by the outermost myelin lamellae of neighboring axons. The intramyelinic splitting occurs at the intraperiod line.
|
Megalencephalic leukoencephalopathy with cysts (MLC) is a genetic infantile-onset disease characterized by macrocephaly and white matter edema due to loss of MLC1 function. Recessive mutations in either MLC1 or GLIALCAM cause the disease. MLC1 is involved in astrocytic volume regulation; GlialCAM ensures the correct membrane localisation of MLC1. Their exact role in brain ion-water homeostasis is only partly defined. We characterized Glialcam-null mice for further studies.
We investigated the consequences of loss of GlialCAM in Glialcam-null mice and compared GlialCAM developmental expression in mice and men. Glialcam-null mice had early-onset megalencephaly and increased brain water content. From 3 weeks, astrocytes were abnormal with swollen processes abutting blood vessels. Concomitantly, progressive white matter vacuolization developed due to intramyelinic edema. Glialcam-null astrocytes showed abolished expression of MLC1, reduced expression of the chloride channel ClC-2 and increased expression and redistribution of the water channel aquaporin4. Expression of other MLC1-interacting proteins and the volume regulated anion channel LRRC8A was unchanged. In mice, GlialCAM expression increased until 3 weeks and then stabilized. In humans, GlialCAM expression was highest in the first 3 years to then decrease and stabilize from approximately 5 years. Glialcam-null mice replicate the early stages of the human disease with early-onset intramyelinic edema. The earliest change is astrocytic swelling, further substantiating that a defect in astrocytic volume regulation is the primary cellular defect in MLC. GlialCAM expression affects expression of MLC1, ClC-2 and aquaporin4, indicating that abnormal interplay between these proteins is a disease mechanism in megalencephalic leukoencephalopathy with cysts. Complete study can be found at: onlinelibrary.wiley.com/doi/full/10.1002/acn3.405 |
First mouse model for human MLC disease. Department of Child Neurology, Neuroscience Campus Amsterdam.
(Annals of Neurology)
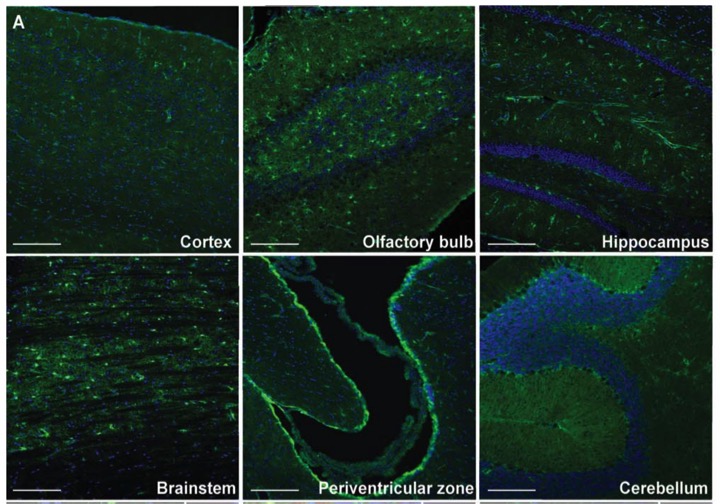
Green fluorescent protein expression tagged to MLC1-promotor in mouse brain areas. In this mouse study, MLC1 protein was replaced with green fluorescent protein. We could see the expression patter of MLC1/eGFP in astrocytes of mouse brain.
|
My PhD project aim is to study a rare genetic childhood white matter leukodystrophy (Megalencephalic leuckoencephalopathy with subcortical cysts; MLC; experiment made by the nature). Patients with MLC develop macrocephaly within the first year of life. After several years, slowly progressive cerebellar ataxia, spasticity and epileptic seizure occurs. Patient MRI reveals swelling of the cerebral white matter and subcortical cycts. Brain biopsies reveal that increase water content is due to the vacuoles in the outer lamellae of the myelin sheaths and astrocytic endfeet. The previous studies have suggested that mutations in the genes encoding the astrocytic membrane protein MLC1 (~ 75% patient pool) or the cell adhesion molecule GLIALCAM (~ 25%; a chaperon protein for MLC1) underlie the pathophysiology of MLC in humans. Building evidence suggests MLC1 as likely link between leukodystrophy and brain channelopathies. The challenge of my project is to understand the mystical pathophysiological association between astrocytic protein and pathology of myelin and observed phenotype of neuronal dysfunction.
|
|
Marjo’s lab developed a MLC mouse model and my first aim was to characterize the mouse model and compare the model with human MLC disease. To characterize the MLC mouse model, I had this opportunity to work with an excellent neuropathologist, Dr Marianna Bugiani. We used multi-disciplinary approach (from molecule to behavior phenotype) to understand the development of the disease in the mice and to study the mouse model in relation to MLC in humans. Aiming to characterize the mouse model, we studied MLC1 protein expression patterns in humans and normal mice. We looked at the development of brain pathology and used different pathological markers at the embryonic aged mice to 12 months old mice. To understand the underlying pathophysiology, I prepared primary cortical astrocyte culture from MLC mice and recorded anion selective currents. In line with previous human astrocytes study, I found reduced outward rectifying volume regulating anion currents in the MLC knockout astrocytes. To rescue the phenotype, I expressed MLC1 construct with the help of adenovirus. Using fluorescent-quenching method, I showed the reduced anion currents leads to impaired volume regulation in MLC knockout astrocytes. In this study, we concluded that MLC mice replicate human MLC disease and MLC1 is the cardinal protein for volume regulation in astrocytes, which underlie the pathophysiology the disease.
The study of MLC mouse model was published in the January 2015 issue of Annals of Neurology together with an image from the study as the front page of the journal. Complete study can be found at: onlinelibrary.wiley.com/doi/10.1002/ana.24307/abstract |
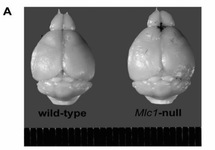
Swollen brain of MLC1 knock-out mouse compared to wild-type mouse (top). Reduced volume regulation anion current together with disturbed regulatory volume decrease in MLC1-KO astrocytes. (red colour is for Mlc1-ko and blue for wild-type animals; Green correspond to aden-virus induced expression of MLC1)
|

Synaptic plasticity modulation in anatomically identified hippocampal inhibitory interneurons. Department of Pharmacology, University of Oxford, The United Kingdom.
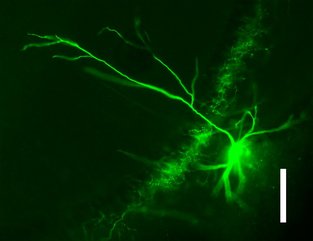
Picture of an Axo-axonic cell from hippocampus of rat brain. While recording these cells, I filled these cells with biocytin and counter stained them with fluorescent marker to morphologically identify the recorded cells.
|
Brain is like an orchestra band with different musical tone from different instruments in a band. Symphony of the different instruments produce a special kind of music or melody. Neuroscientist are trying to understand such codes of rhythms that are involved in defining our thoughts, memory and abilities to perform complex task. Each instrument in our brain can correspond to a specific brain cell (Neuron) which creates a special rhythm or electrical firing (action potential) pattern. In our brain majority of the cells are excitatory (Have excitatory neurotransmitters; Depolarise the cell) and minority of inhibitory interneurons (release inhibitory neurotransmitters; hyperpolarize the cell). Although Interneurons are minority in the brain, these cells play the central role in setting the rhythms of firing patterns in the brain.
|
|
I was accepted by Dr Karri Lamsa at Department of Pharmacology, Oxford, to learn patch clamp recordings from acute brain slices from young rats. My focus at Karri’s lab was to record the role of nicotinic receptors on synaptic plasticity of morphologically identified interneurons in hippocampal CA1. In this project, I learned the whole cell current clamp recording using Axoclamp-2B amplifier and post hoc analysis of the recorded cells. I recorded 46 O-LM (Oriens Lacunosum Moleculare) cells and 6 Axo-axonic cells at Karri’s lab. I showed nicotine facilitates long-term plasticity in O-LM cells when combined with high frequency stimulation pulse. A confocal image of the recoded O-LM cell was selected by the department pharmacology as the front page of the website. I also presented this work as a poster presentation at the OXION-2010 (Oxford-Cambridge-UCL combined initiative for Ion channels in excitable membrane).
|
Screen picture of the Department of Pharmacology, Oxford website. Confocal image of the soma of O-LM cell stained with somatostatin.
|
Deep brain stimulation; A hope for drug resistant depression patients. Department of Pharmacology, University of Oxford, the United Kingdom.
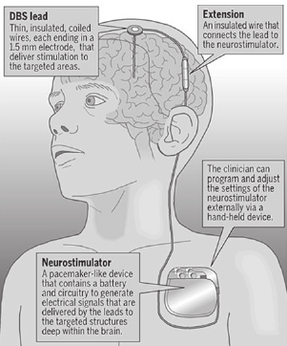
Image shows the Deep Brain Stimulation setup. Brain electrodes are implanted in the required location by the neurosurgeon. Stimulator can be placed in satellite location in the body. Above setup is ideal for Parkinson patient where periodic stimulation can be required to subsidies disease symptoms. This image is take from cnsdisease.com.
|
Major depression is a commonly occurring, serious, recurrent disorder linked to diminished role functioning and quality of life, medical morbidity, and mortality. The World Health Organization (WHO) has ranked depression the 4th leading cause of disability worldwide and projects that by 2020, it will be the second leading cause. Another challenge is to handle the cases where conventional antidepressant drugs fail to be the answer. In this project, I did literature investigation for deep brain stimulation technique as a treatment option for drug resistant depression. Deep brain stimulation (DBS) is a technique where specific part of the brain is electrically stimulated with multiple electrodes to relieve patient symptoms. Currently, DBS is FDA approved technique for parkinson patients. My motivation for opting this project was after reading a clinical case study where a female parkinson patient showed depression like symptoms after DBS. I investigated the areas of the brain that could be effective target for stimulation in depressed patients. I received distinction for essay writing including this project. I performed literature research under the supervision of Professor Trevor Sharp at Department of Pharmacology, University of Oxford.
|
Corporate Social Responsibility; Case study on Ranbaxy Laboratory LTD, Delhi, INDIA.
|
Any non-governmental corporation is generating revenue from the public directly or from the public funds. I always feel it is the ethical and moral duties of the big corporations to be involved in significant philanthropic activities. During my final year of pharmaceutical studies, I decided to make a case study on one of the big pharmaceutical company in India, Ranbaxy Laboratories LTD. I looked up at the philanthropic activities performed by the company in year 2008.
|

|
Pharmacology project; Department of Pharmacology, Maulana Azad Medical College, Delhi, INDIA.

|
Each medicine that can be given to human beings is suppose to pass through a preliminary safety screen of pre-clinical animal research. Recent scientific world has given immense number of life saving drugs to human kind and it has only been possible due to the dedicated research of multiple scientist and there work on animals models. Preclinical animal research is not only vital but indispensable for certain type of research questions. Hence, it is important to first learn how to handle and make use of animals efficiently to answer given research question. I spent six months in department of pharmacology, Maulana Azad Medical College, Delhi, studying various ways of animal handing in laboratory together with pre-clinical investigation of certain under research anti-epileptic medicines. This work was supervised by Dr Somnath Datta and Prof Uma Tekur (Department Head, Pharmacology, MAMC).
|
Ranbaxy production unit; Ponta Sahib, Himachal Pradesh, INDIA
|
Once scientist manage to find a new medicine for any specific disease the next challenge is to deliver the drug to the right place with correct strength. The science of developing drug into its dosage form is called Pharmaceutics. In 2006, I spent summer training to understand and learn how to develop Tablets, Soft gelatine capsules and sustain release dosage forms at "Ranbaxy Laboratories LTD". I also learned how to screen the active pharmaceutical ingredient in the final dosage form using multiple spectroscopic and chromatographic analysis. Designing smart medicine is one of the most challenging task for pharmaceutical scientist. How to send a correct medicine to the brain across the brain fluid barrier is a daunting task. Novel drug delivery system is always my second love after Neuroscience. Picture taken at the entrance of the core-zone of the production unit. You have to wear another suite before you can enter the core zone (under GMP guidelines). Core zone is where actual medicine (Solid, Liquid or vapours) is punched into tablets or placed into soft gelatine capsules.
|

|
Model of New Era (2001-2002)

|

Together with Bhanu Minocha, I was jointly awarded with outstanding science award at the school, district, zone, state (NCT-Delhi) and finally at the National science exhibition (2001-2002). At these exhibitions, we presented an earthquake proof 20th century multistory building model. The requisite to build such model came from a significant impact of a massive earthquake that took thousands of lives on 26th January 2001 in northwest India. We applied simple understanding of high school physics and used it in the project. We used centre of gravity, air-gap suspension mechanism as the key concept for the model. During the presentations, our project received attention from National Physical Laboratory, Delhi and Indian Space Research Organization scientists. Pictures are from Jawahar Lal Nehru National Science Exhibition 2002, organised by National Council for Education Research and Training (NCERT) held at Gandhi Bhawan, Hydrabad, Andrapradesh, INDIA. This work was also published by NCERT. In our high school, we were the "Earthquake boys" shaking the ground with our ideas! (Bhanu graduated as Engineer and currently Project Manager in a ACME Solar Energy company in Delhi, India)
|